The Tabas laboratory studies the cellular biology of cardiometabolic disease, with an emphasis on the molecular-cellular mechanisms of advanced atherosclerosis and hepatic insulin resistance and NASH in obesity, and the links between these processes.
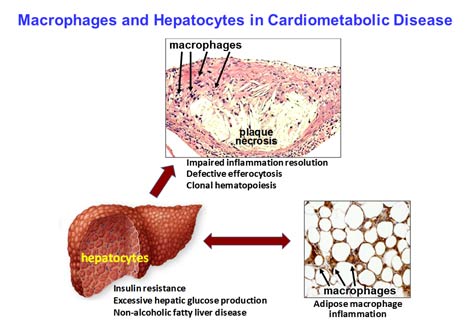
The studies on advanced atherosclerosis have focused on integrated processes that combine to promote advanced plaque progression, with a focus on defective clearance of apoptotic cells (efferocytosis) and impaired inflammation resolution. In the efferocytosis field, current studies are focused on (a) cell biological pathways triggered by macrophage metabolism of ingested and degraded apoptotic cells, particularly pathways related to inflammation resolution; and (b) linking macrophages processes that promote atherosclerosis progression to both human genetic risk factors for coronary artery disease, i.e., polymorphisms that regulate specific genes, as well as systemic insulin resistance. The inflammation resolution project has a strong focus on processes that promote plaque regression and on the therapeutic potential of promoting resolution, e.g., through the use of atherosclerosis-targeted nanoparticles packaged with resolution mediators. Additionally, an exciting new area in the lab involves identifying specific mechanistic links between impaired resolution pathways and clonal hematopoiesis of indeterminate potential (CHIP), which is a major risk factor for atherosclerosis and coronary artery disease in humans over the age of 60.
The lab's studies on hepatic insulin resistance, which is an important driver of atherosclerosis, led to the discovery of a new calcium-stimulated pathway in hepatocytes that plays a key role in glucagon-mediated excessive glucose production, insulin resistance, fatty liver, dyslipidemia, impaired thrombolysis, and adipose tissue inflammation in the setting of obesity and type 2 diabetes. Ongoing studies are investigating (a) how the pathway may promote atherosclerotic plaque progression by affecting plaque macrophages; and (b) strategies to translate our discoveries into new types of drugs to treat type 2 diabetes and prevent diabetes-driven atherosclerotic plaque progression.
Related to the lab's metabolism work, we study hepatocyte pathways in non-alcoholic steatohepatitis (NASH), with current investigations focused on (a) the role of a transcription factor called TAZ in both NASH fibrosis and NASH-associated hepatocellular carcinoma; (b) new mechanistic insight into known human genetic risk factors for NASH fibrosis; and (c) new therapeutic strategies to prevent steatosis-to-NASH progression based on our findings.
The overall approach of the laboratory is to elucidate in-depth mechanisms using molecular-cell biological approaches; test causation in normal physiology and disease models using genetically engineered mice; probe relevance to humans through the study of normal and diseased human tissues and human genetics; and conceive and test novel and mechanism-based therapies in the cardiometabolic arena. Our human genetics studies are being conducted in collaboration with the CUMC Cardiovascular and Metabolic Precision Medicine Program, led by Dr. Muredach Reilly, and include plans to study human iPSC-derived macrophages that can be genetically engineered using CRISPR/Cas9 to mimic or "correct" disease-causing human mutations. We also have collaborations with Dr. Danish Saleheen at Columbia, who directs a translational program studying humans who have heterozygous and homozygous null mutations in thousands of individual genes.
The laboratory's philosophy is one of collaboration, not competition, and emphasis is placed on long-term, in-depth, and rigorous studies. Dr. Tabas places great emphasis on mentoring, particularly fostering independence, and career guidance. His trainees have had great success in career development grants from the NIH (K99/R00) and American Heart Association and subsequently with NIH R01 grants soon after leaving the lab for independent tenure-track Assistant Professorships at major university research organizations. |